Coarse-Graining
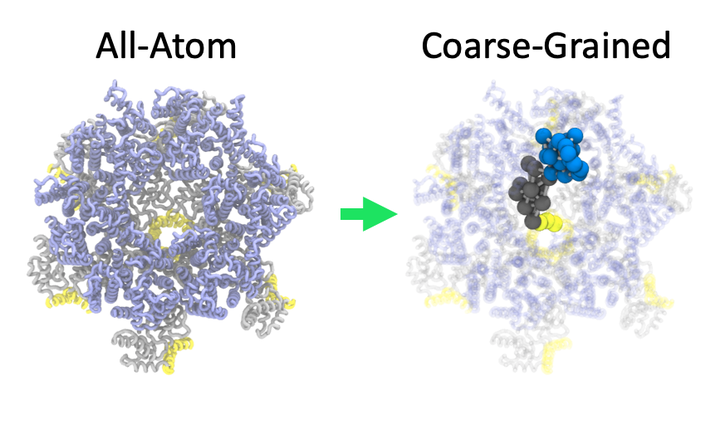 Coarse-graining of the immature HIV-1 CA/SP1 protein
Coarse-graining of the immature HIV-1 CA/SP1 proteinWe study how to effectively model macromolecules at reduced resolution to faciliate dynamical simulations at longer and larger scales.
Why is this useful? The hierarchical and dynamical nature of macromolecular materials necessarily involve physics over a range of length and time scales, e.g. from a few atoms when studying ligand binding (e.g. small-molecule drugs or biosensor analytes) to billions of atoms when understanding how proteins assemble into functional macromolecular machines. Simulating these systems remains out of reach to modern atomistic molecular simulation algorithms. Our strategy is to model these systems by removing rapidly-changing degrees of freedom through coarse-graining. We develop systematic techniques using statistical mechanics to derive effective coarse-grained interactions. These models allow us to explore inherently large and/or slow collective phenomena during the assembly and reorganization of macromolecular complexes.
Alexander J Pak
Assistant Professor of Chemical and Biological Engineering
My research interests include molecular simulations, coarse-graining, soft matter, self-assembly, and bio-inspired materials